The Vaxign Tutorial
Notes:
- The following tutorial comes from the Vaxign Demo material prepared for the ICoVax 2012 Workshop.
- This material was prepared by Allen Xiang and Oliver He and was finalized on October 6, 2012.
- The PDF copy of the material is available HERE.
Introduction. Vaxign is a web-based software program in VIOLIN that targets for vaccine design. Based on the reverse vaccinology strategy, Vaxign predicts vaccine targets by bioinformatics analysis of genome sequences. The genome sequences come from pathogenic strains, non-pathogenic strains (optional), and host species (human, mouse, or pig). Predicted features in the Vaxign pipeline include protein subcellular location, transmembrane helices, adhesin probability, conservation among pathogenic strains, sequence exclusion from genomes of nonpathogenic strains, sequence similarity to host proteins, and epitope binding to MHC class I and class II. Vaxign contains precomputed predictions for over 200 genomes and also allows dynamic vaccine target prediction based on users’ input sequences.
The following tutorial will provide step-by-step instructions on how to navigate through the Vaxign website (https://violinet.org/vaxign). Please feel free to ask any questions throughout this training exercise. Two use cases will be tested: (1) Human Herpesvirus 1 (HSV-1) vaccine target prediction; (2) Enterohemorrhagic Escherichia coli (EHEC) O157:H7 vaccine target prediction. The first HSV use case is mainly for demonstrating Vaxign sequence conservation analysis and MHC class I epitope prediction techniques. The O157:H7 use case is mainly for showing some general features for bacterial vaccine target prediction, such as the prediction of secreted and outer membrane proteins and transmembrane helices. To save time, we will mainly use the pre-computed results. We will also go through how to set up your own account in Vaxign for your analyzed data storage and collaboration with others.
FIRST USE CASE: HSV-1 VACCINE TARGET PREDICTION
Enter Vaxign web page. Open a browser, such as Firefox or Internet Explorer. Type the URL: http://violinet.org/vaxign. Then you will come to the Vaxign cover page (Figure 1). Vaxign is a software program freely available for public uses. You do not need to sign any license or log in in order to use it. It is noted that we do provide an optional log in service if you want to save your analyzed data for personal or collaborative uses. This feature will be excised later. There are two methods for running Vaxign: one is Vaxign query for pre-computed results, the other is dynamic analysis for your own sequence input. We will mainly focus on the Vaxign query method (Figure 2).
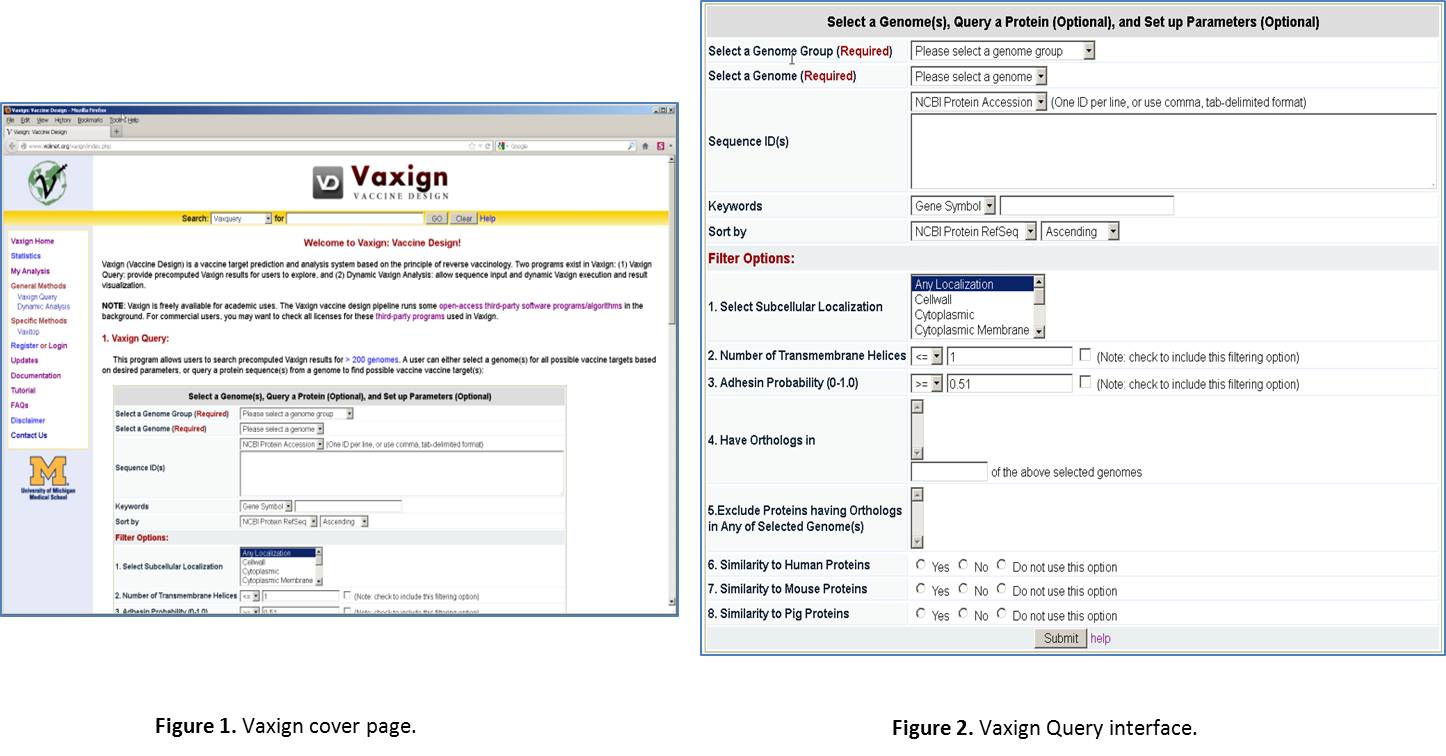
Select a Genome Group. In Vaxign Query site, click the “Please select a genome group” dropdown list. Select the option “Herpesvirus (52)”. The number “52” means that this group contains 52 genomes, which are from 52 herpesvirus strains.
Select a Genome. From the drop down list, select “Human herpesvirus 1”.
Ignore “Sequence ID(s)” line. This part is for querying one or a list of sequence IDs from the specified genome. The IDs can be NCBI Protein Accession number, NCBI Protein GI, NCBI Gene ID, and NCBI Locus Tag.
Ignore “Keywords” line. This part is for querying one or few proteins based on a specific database ID (e.g., NCBI Locus Tag), gene symbol, or protein description.
Choose the default for “Sort by”. This setting is for how you want to view the results. It is fine to just stick with the default.
Select Filter Options. We have a list of options for you to filter out proteins. Here is a brief introduction of different options:
- Select Subcellular Localization. This feature does not particularly fit in this virus use case. It is useful for bacterial vaccine target prediction. For bacterial vaccine development, it is often preferred to identify outer membrane or secreted proteins as vaccine targets.
- Maximum Number of Transmembrane Helices. It has been observed that a protein with more than one transmembrance helix is hard to be isolated from a recombinant E. coli strain. Therefore, this feature is useful if you consider using recombinant E. coli strains for protein isolation and purification.
- Minimum Adhesin Probability (0-1.0). An adhesin is a protein critical for helping a pathogen to enter a host cell. Neutralizing an adhesin is helpful for preventing a pathogen’s invasion. The default cutoff is 0.51.
- Have Orthologs in. This feature is used for finding conserved proteins among a selected list of strains. Tips: To select (or unselect) individual strains, press Ctrl key and then select; For strain selection in continuous order, you can also use Shift key. In the end of the selection box, you can type a number “of the above selected genomes”. This number can be any integer number equal to or less than the maximum number of selected genomes. If you do not type any number, the default is the total number of selected genome. This definition allows you to look for those orthologs that appears in all or part of the selected genomes.
- Exclude Proteins having Orthologs in Any of Selected Genome(s). This is for excluding proteins that also also exist in a non-pathogenic strain(s).
- No Similarity to Human Proteins. Choose this selection to exclude those vaccine targets that also exist in humans.
- No Similarity to Mouse Proteins. Choose this selection to exclude those vaccine targets that also exist in mouse.
- No Similarity to Pig Proteins. Choose this selection to exclude those vaccine targets that also exist in pigs.
NOTE: MHC Class I & II Epitope Prediction by Vaxitop. This option has been removed from current version of Vaxign query cover page. However, you can use the separate Vaxitop program to run it (URL: https://violinet.org/vaxign/vaxitop). Also, although the Vaxitop epitope prediction selection is not available in the cover page, the Vaxitop epitope prediction method is available for any protein you select after the initial screening. To predict MHC class I and II epitopes, the Vaxitop method is based on prediction of position specific scoring matrices (PSSM). Different from existing epitope prediction algorithms, Vaxitop relies on statistical P-value (instead of a percentage or top number) as the cutoff. A P-value of 0.05 provides a cutoff with high and balanced sensitivity and specificity. Under this section, you can choose a P-value cutoff, host species, MHC allele, and epitope length. It’s noted that the Vaxitop name replaces previously defined “Vaxitope” in order to avoid a naming conflict.
To illustrate step-by-step how Vaxign works, for now let’s NOT select any filter option.
Submit your query. Slick the “Submit” button in the bottom of the Vaxign Query web interface.
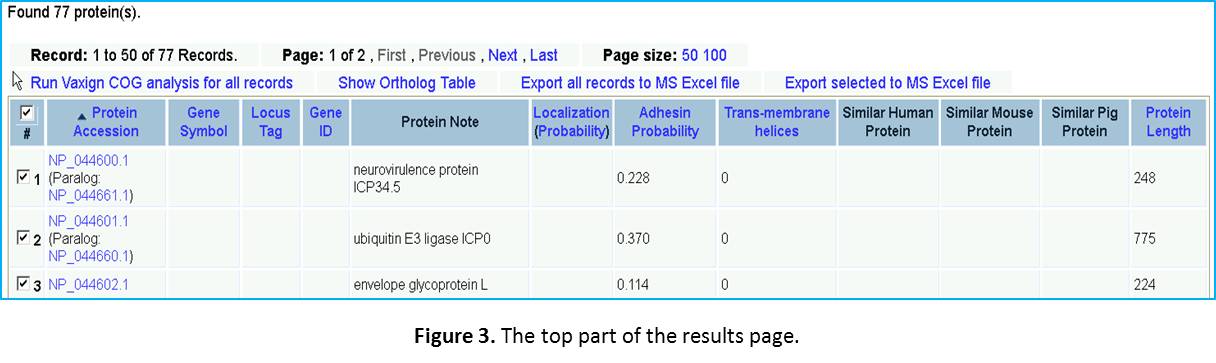
Query result examination. While a little while, you will come to a Vaxign query result display page. You will see the first line saying “Found 77 protein(s).” The top part of the results web page is copied in Figure 3. As you can see here, only the first 50 of the 77 proteins are display on this page. You can click “Next” or “Last” to find the information for the remaining 27 proteins. If you prefer, you can also change the Page size to 100 to show all the 77 proteins in one page. Each row of the table is the information for a specific protein, which includes: (1) Protein Accession number, (2) Gene Symbol, (3) Localization, (4) Adhesin Probability, (5) Trans-membrane helices, (6) MHC class I binding epitopes, (7) MHC class II binding epitopes, (6) Similar human proteins, (7) Similar mouse proteins, and (8) Similar pig proteins.
Conservation among herpesviruses. We will now identify how many proteins are conserved in other human herpesviruses.
- Conservation among two other HSV-1 genomes.
- Scroll down your web page to the setting of “Have Orthologs in”, select “Human herpesvirus 1 strain F” and “Human herpesvirus 1 strain H129”. Scroll down a little more to under the selection box, type “2” before the phase “of the above selected 2 genomes”. If you do not type anything, the default is that you choose the total number of selected genomes. Note: if you type 1, it means that you want your resulted protein to have conservation in at least one selected genome.
- Scroll down and click “Search” at the bottom of the page.
- Results. You will come to another page. This page indicates still 77 proteins. This means that all 77 proteins are conserved in all three HSV-1 strains.
- Conservation among 11 other HSV genomes.
- Instead of choosing only the above 2 HSV-2 genomes, now select all 11 human herpesviruses. Scroll down a little more to under the selection box, type “11” before the phase “of the above selected 11 genomes”.
- Scroll down and click “Search” at the bottom of the page.
- Results. You will come to another page. Now you will find only 19 proteins. This means that only these 19 proteins are conserved in all 12 HSV strains (i.e., 11 + 1 seed strain).
Restriction of maximum number of transmembrane helices. Now we want to restrict the maximum number of transmembrane helices to one.
- Scroll down to the setting “2. Maximum Number of Transmembrane Helices”. Type “1” (or leave it if it is already 1) and select this option.
- Scroll down and click “Search” at the bottom of the page.
- Results. You will come to another page. Now you will find only 17 proteins. This means that two proteins have been filtered out.
Sort results by Adhesin Probability. Now we will excise the adhesin probability prediction.
- Click the table header “Adhesin Probability”. This will sort the 17 proteins based on the adhesin probability.
- Results. Only one protein “NP_044628.1” (a capsid scaffold protein) has an adhesin probability higher than 0.51. The adhesin probability of this protein is 0.675. This is like that this protein is an adhesin protein (Note: There appears no literature report that confirms this prediction.)
Checking detailed results for one protein. Now we will examine one protein in details.
- Click the protein “NP_044628.1”. Then we will come to a page with detailed predictions for this protein. Parts of the results are shown in Figure 4.
- Alpha Helix Prediction. Click on the “Alpha Helix Prediction” on this page. The result of predicted alpha helix for this protein will be shown on a new page. No transmembrane domain is found. It appears that this protein is secreted out of a cell.
- MHC Class I Epitope prediction. Next we will exercise to find out specific MHC class I epitopes for this protein. Note that we will not work on MHC class II epitope prediction in this exercise.

- Human MHC Class I Epitope prediction. We will focus on prediction of human MHC class I epitopes.
- P-value Cutoff. Choose the default 0.05.
- MHC Host Species. Choose “human” in the dropdown list.
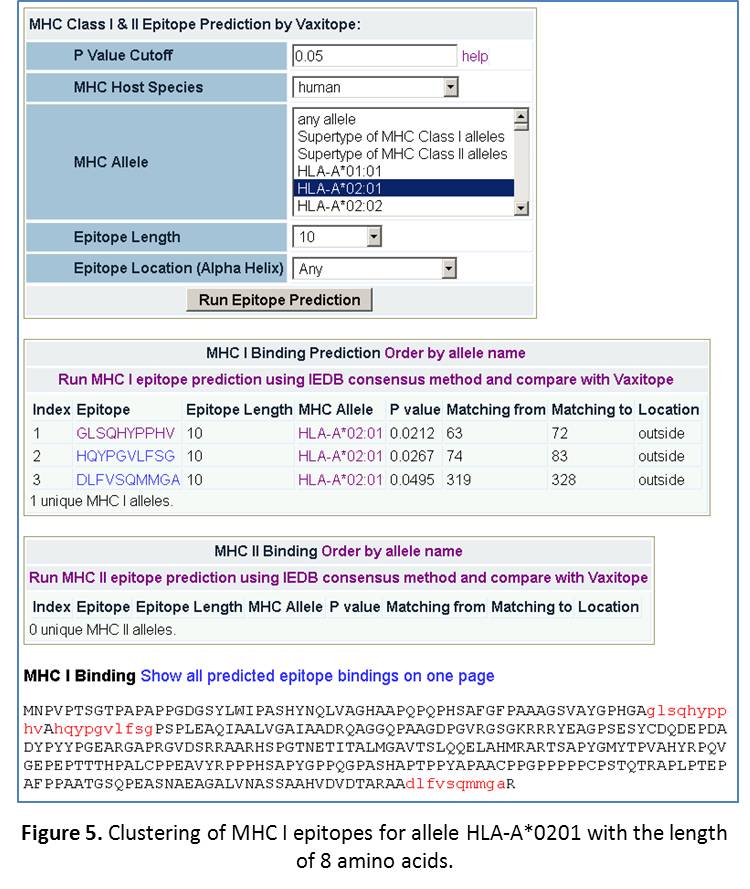
- Predict epitopes for a MHC Allele with any epitope length. If we choose “any allele” and any epitope length, we will get 484 MHC class I epitopes for 53 unique MHC I alleles, and 198 MHC class II epitopes for 36 unique MHC II alleles. If we choose one common allele “HLA-A*0201” and length “10”, we will get 3 MHC class I epitopes for this allele with a length of 10 amino acids. These epitopes are distributed in different areas of the protein. Scroll down to the bottom of the page, you can see the clusters of these epitopes inside the protein (Figure 5).
- Predict epitopes for a specific allele with a specific epitope length. Now, select the setting: human HLA-A*0201, epitope length: 10. Click “Refresh”. The results are shown in Figure 5. Basically, we get three hits. The Vaxitop P-values for them are 0.00212, 0.0267, and 0.0495. The positions for each of the epitopes are also displayed (Figure 5).
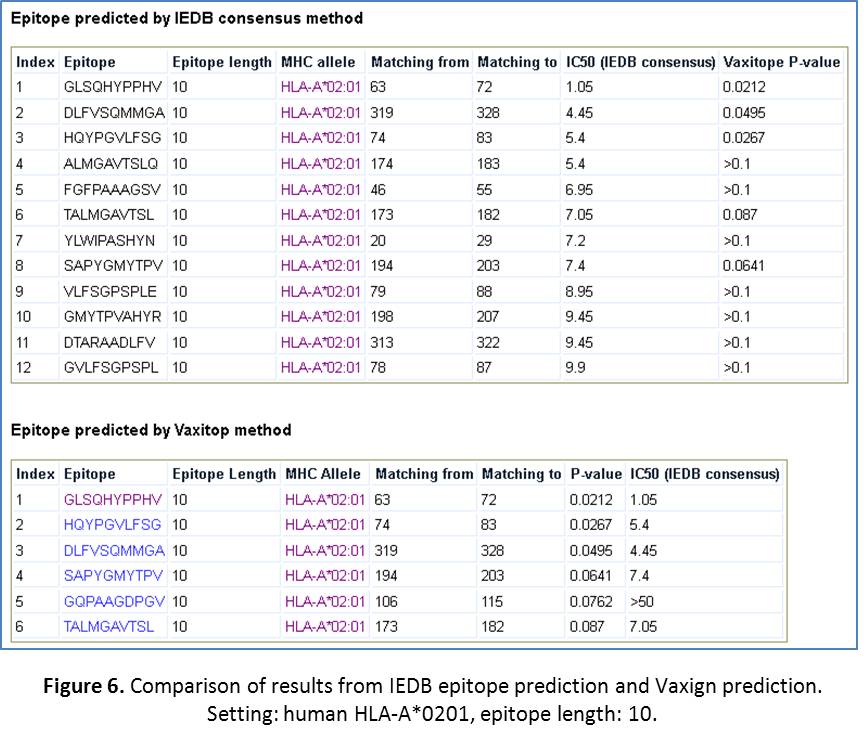
- Compare Vaxign epitope prediction with IEDB epitope prediction. The IEDB MHC class I epitope prediction method is a well-known and commonly used method (URL: http://tools.immuneepitope.org/analyze/html/mhc_binding.html). We have downloaded a copy of the IEDB MHC class I epitope prediction software and installed it in Vaxign for specific comparison with our own method. The commonly used IEDB MHC class I “consensus” prediction method is used for this purpose. To run a comparison, click on “Run MHC I epitope prediction using IEDB consensus method and compare with Vaxitop”. After the click, you will come to a page that you can select the MHC allele HLA A*0201 and the length of 10. Then click “Submit”. The results are shown in Figure 6. It appears that the top three results out of the two methods are the same three epitopes – the best one with Vaxitop P-value of 0.00212. The IEDB method turns to provide more positive results using their IC50 cuttof of 50. We usually provide their positive results here with the IC50 cutoff value of 10. Even with this cutoff, many positive values were predicted using the IEDB method but not with Vaxitop (Figure 6). Overall, there is an overlap and also difference between these two methods. One possible solution in practice is to look for the shared results out of these two methods.
Gene function grouping and annotation. After selection of a list of genes, the results can be automatically exported to DAVID for functional annotation and grouping. We have also an internally developed COG analysis to cluster selected genes for COG functional analysis. These two features are available by clicking corresponding links immediately after the query result table.
Export results. The predicted results can be exported to MS Excel file.
SECOND USE CASE: E. COLI O157:H7 VACCINE TARGET PREDICTION
We will not provide much detail for this use case here. The procedure to run this is similar to what we explain above. One feature that is not demonstrated in the above example is the Filter Option:
- 1. Select Subcellular Localization. This option allows you to select one or more subcellular locations. The options include:
- Any Localization
- Cell wall
- Cytoplasmic
- Cytoplasmic Membrane
- Excellular proteins
- Outer Membrane
- Periplasmic
- Unknown.
It is noted that this filter method is based on PSortb (http://www.psort.org/psortb/), which is specifically designed for prediction of subcellular localization of proteins from Gram + or Gram – bacteria. It is not designed for other types of microbes.
Figure 7 provides some concrete settings for an example analysis. In this example, we chose to identify only those extracellular and outer membrane proteins. This analysis results in 36 hits.
Register and Use a Vaxign Account for Predicted Result Storage and Sharing
Purpose of having a Vaxign account. It is not required to open a Vaxign/VIOLIN account. However, it provides some extra benefits for you: (1) It saves your dynamic analysis results in the Vaxign system for your future reference and refinement. (2) It promotes collaboration. You can share your Vaxign projects with your colleagues, so the whole team can work on one Vaxign analysis project together.
Set up a Vaxign account. Click “My Analysis” on the left side navigation bar in the Vaxign system (Figure 8). If you have not register, please click on “Register an account” and register using a web form.
Create a new project under “My Analysis”. You can create a new project and follow the online instruction.
Exit Vaxign. Once you complete your exercises, you may log out of Vaxign if you generate your own account and log in. To log out, navigate to the top of the window, and click on LOGOUT.
------------------------
Note: Please see more information in our Vaxign Documentation page.
Provide Comments. Once you complete your exercises, you may log out of Vaxign if you generate your own account and log in. In an effort to improve the Vaxign performance and provide better support for the vaccine development community, we would appreciate any comments/suggestions you may have regarding the Vaxign program. To provide your comments, please email us or fill out a feedback online form in the Vaxign Contact Us page. Thank you!!


