|
|
The Vaxign Documentation
Note: Vaxign and Yongqun "Oliver" He have been cited in news: Reverse vaccinology on the cusp by Dan Jones. Nat Rev Drug Discov. 2012 Feb 10;11(3):175-6. doi: 10.1038/nrd3679. PMID: 22322255. Please click HERE to find more papers that have cited Vaxign.
In the post-genomic era, strategies of vaccine development have progressed dramatically from traditional Pasteur’s principles of isolating, inactivating and injecting the causative agent of an infectious disease, to reverse vaccinology that starts from bioinformatics analysis of the genome information. Vaxign is the first web-based vaccine design software program freely available for the purpose of facilitating reverse vaccinology. Here we provide some relevant documentation about Vaxign:
Table of Contents:
- Vaxign Publications
- Vaxign Pipeline for Vaccine Target Prediction
- Vaxign-ML Pipeline for machine learning-based Vaccine Target Prediction
- Open-Source Software Programs/Databases used in Vaxign and their Licenses
- Parameters/Options for Consideration and Filteration
- Introduction of Vaxitop
- Selected Relevant References
- Selected Papers that have Cited Vaxign
- Useful web links
--------------------------------------------------------
Vaxign Publications:
He Y, Xiang Z, Mobley HLT. Vaxign: the first web-based vaccine design program for reverse vaccinology and an application for vaccine development. Journal of Biomedicine and Biotechnology. Volume 2010 (2010), Article ID 297505, 15 pages. [PMID: 20671958] (Note: this paper introduces the Vaxign program and a use case of how to use Vaxign for uropathogenic E. coli vaccine target prediction. Please use this paper as your formal Vaxign citation.)
Xiang Z, He Y. Genome-wide prediction of vaccine targets for human herpes simplex viruses using Vaxign reverse vaccinology. BMC Bioinformatics. 2013, 14(Suppl 4):S2 (8 March 2013). PMID: 23514126; PMCID: PMC3599071. (Note: this paper introduces some progresses made in Vaxign up to the end of 2012. You can also use this paper as your formal Vaxign citation.)
Xiang Z, He Y. 2009. Vaxign: a web-based vaccine target design program for reverse vaccinology. Procedia in Vaccinology. Volume 1, Issue 1, Pages 23-29. (Note: this paper was presented in the ISV vaccine meeting in 2008. It first introduces our Vaxign program and provides some use case study results.)
He Y, Xiang Z. Bioinformatics analysis of Brucella vaccines and vaccine targets using VIOLIN. Immunome Research. 2010 Sep 27;6 Suppl 1:S5. PMID: 20875156. PMCID: PMC2946783. (Note: this paper introduces a use case of how to use Vaxign for Brucella vaccine target prediction)
He Y. Genome-based computational vaccine discovery by reverse vaccinology (Chapter 5); in book: Immunomic Discovery of Adjuvants and Candidate Subunit Vaccines, edited by Darren R. Flower, Yvonne Perrie. Publisher: Springer, New York, 2013. ISBN-13: 978-1461450696. ISBN 978-1-4614-5070-2 (eBook). Links in: Google eBook, and Amazon book.
Papers that cited Vaxign through collaboration with the Vaxign team:
McNamara L, He Y, Yang Z. Using epitope predictions to evaluate efficacy and population coverage of the Mtb72f vaccine for tuberculosis. BMC Immunology. 2010 Mar 30;11(1):18. [PMID: 20353587] [Journal Link]
Ma J, He Y, Hu B, Luo ZQ. Genome sequence of an environmental isolate of the bacterial pathogen Legionella pneumophila. Genome Announc. 2013 Jun 27;1(3). PMID: 23792742. PMCID: PMC3675512.
More papers that have cited Vaxign. Please check HERE.
Vaxign Pipeline for Vaccine Target Prediction:
Vaxign includes a pipeline of software programs to predict possible vaccine targets based on various vaccine design criteria using microbial genomic and protein sequences as input data. The predicted features in the Vaxign pipeline include antigen sublocation, adhesion, epitope binding to MHC class I and class II, and sequence similarities to human, mouse and/or pig proteins. This pipeline integrates both existing open source tools and an internally developed program (Vaxitope) with user-friendly web interfaces. Vaxign predicts vaccine targets based on protein sequences at a genome level or using individual protein sequences. This pipeline includes the following steps:
- Subcellular localization: Surface-exposed proteins such as outer membrane proteins (esp. adhesins) and secreted proteins are usually ideal targets for vaccine developments. Non-surface proteins such as cytoplasmic/inner membrane proteins may not be good targets for vaccine development.
- Topology and Transmembrane helices: It is very difficult to clone, express, and purify proteins with more than one transmembrane spanning region. Therefore, it might be better to ignore those proteins with multiple transmembrane spaaning regions in the first place.
- Adhesin probability: Adhesins are often good vaccine targets.
- Epitope prediction: This step predicts both MHC class I and class II binding epitopes using Vaxitope, an internally developed program.
- Similarity to host genome sequences: A vaccine candidate with similar sequence to the host (e.g., human, mouse, pig) is likely to cause autoimmunity in the host.
Vaxign-ML Pipeline for machine learning-baesd Vaccine Target Prediction:
Vaxign-ML is a supervised machine learning classification to predict protective antigens. To identify the best machine learning method with optimized conditions, 5 machine learning algorithms (logistic regression, support vector machine, k-nearest neighbors, random forest, and extreme gradient boosting) were tested with biological and physiochemical features extracted from the Protegen database. Nested five-fold cross-validation and leave-one-pathogen-out validation were used to ensure unbiased performance assessment and the capability to predict vaccine candidates for a new emerging pathogen. The best performing model, Vaxign-ML (extreme gradient boosting trained on all Protegen data), was compared to three publicly available reverse vaccinology programs with a high-quality benchmark dataset, and showed superior performance in predicting protective antigens.
(Paper in preparation)
Vaxign-ML standalone version is available in Docker. Source code is avaiable in GitHub
Installation:(Docker version >=1.13.1, API version >=1.26)$ docker pull e4ong1031/vaxign-ml:v1.0$ wget https://raw.githubusercontent.com/VIOLINet/Vaxign-ML-docker/master/VaxignML.sh$ chmod a+x VaxignML.sh$ ./VaxignML.sh [INPUT_FASTA] [OUTPUT_DIRECTORY] [ORGANISM_TYPE](You may need root privilege to run docker commands)
Open-Source Software Programs/Databases used in Vaxign and their Licenses:
- PSORTb: PSORTb (v.2.0) is probably the most precise bacterial localization prediction tool. PSORTb is under the GNU General Public Licence (GNU GPL).
- TMHMM: Prediction of transmembrane helices in proteins. TMHMM is uder Academic Software License Agreement.
- SPAAN: Prediction of adhesins and adhesin-like proteins. According to the SPAAN publication, the SPAAN program is freely available.
- BLAST: NCBI sequence similarity alignment and analysis program. BLAST is a USA NCBI program within the public domain.
- IEDB: The Immune Epitope Database and Analysis Resource. The immune epitope data obtained from IEDB is used for training Vaxitope, our epitope prediction program. Please check the IEDB Term of Use.
Parameters/Options for Consideration and Filteration:
Our automatic Vaxign pipeline allows users to select and/or modify the following parameters:
- Subcellular localization: Please select the localizations you wish to include. Default setting includes (1) Cell Wall, (2) Cytoplasmic, (3) Cytoplasmic Membrane, (4) Extracellular, (5) Out Membrane, (6) Periplasmic, (7) Unknown, and (8) Any Localization (default choice). Please see more details in the PSORTb help page.
- Transmembrane helices: Please enter maximum number of transmembrane helices. Default value is 1 (link to TMHMM help page).
- Adhesin probability: Please specify the minimum value of adhesin. Default value is 0.51 (Sachdeva et al.).
- No similarity to human proteins: Check this option if you wish to exclude any protein that shows any similarity to a human protein.
- No similarity to mouse proteins: Check this option if you wish to exclude any protein that shows any similarity to a mouse protein.
- No similarity to pig proteins: Check this option if you wish to exclude any protein that shows any similarity to a pig protein.
Introduction of Vaxitop:
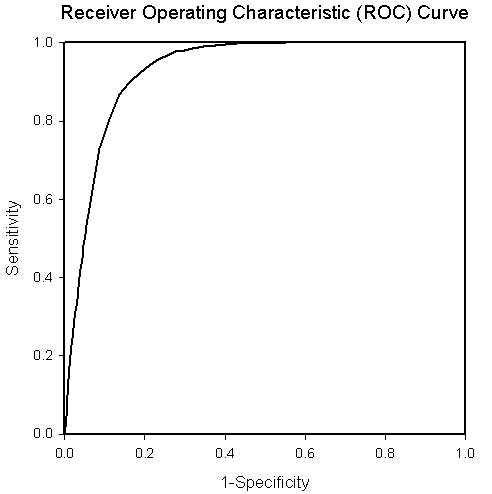
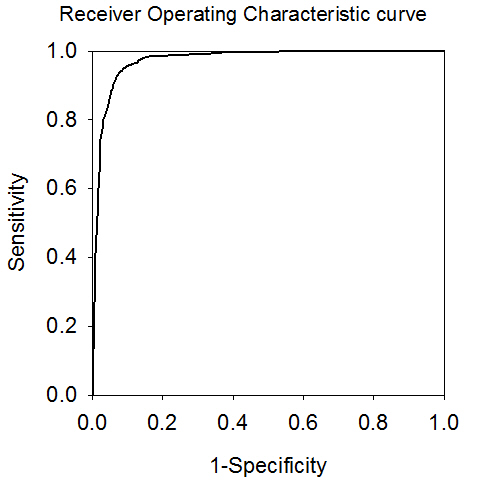
Vaxitop (previously named Vaxitope, now changed to Vaxitop to avoid name conflict) e is an MHC Class I and II binding epitope prediction tool developed in Dr. Yonggqun "Oliver" He's laboratory. Vaxitope is a position specific scoring matrice (PSSM)-based epitope prediction program. Vaxitop relies on statistical P value (instead of a percentage or top number) as the cutoff. Our studies indicate that the P value of 0.05 provides a cutoff with high and balanced sensitivity and specificity. Vaxitop also allows genome-wide query on different MHC host species. To evaluate the performance of Vaxitope, a receiver operating characteristic (ROC) curve was generated using HLA A*0201 specific PSSM. The result is shown below. The value of the Area Under the ROC Curve (AUC) of 0.929 for predicting the epitopes for the allele HLA A*0201 with the length of 9 amino acids. The positive and negative testing dataset was obtained from IEDB. The positive HLA A*0201 aelle epitopes were used to calculate the True Positive Rate (Sensitivity). The negative peptides for the allele with the same lenghth were used to calculate the False Postive Rate (1-Specificity). We have recently updated our program. The performance of our 2012 version has been improved compared to the original 2009 version (as shown below). In addition, we have installed IEDB MHC Class I and II epitope prediciton programs in our systems, and allow a user to compare our Vaxitop-predicted results with the results from the IEDB tools.
| AUC=0.929 (2009 data) |
AUC=0.971 (2012 data) |
 |
 |
The ROC AUCs for predicting epitopes specific for other alleles with different lengths are also calculated and provided for users' reference. Overall, Vaxitop is a very specific and sensitive method for both MHC Class I and II binding epitope prediction.
Selected References:
- Rappuoli R. Reverse vaccinology. Curr Opin Microbiol. 2000 Oct;3(5):445-50. Review. PMID: 11050440.
- Gardy JL, Laird MR, Chen F, Rey S, Walsh CJ, Ester M, Brinkman FS. PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatic. 2005 Mar 1;21(5):617-23. PMID: 15501914.
- Käll L, Krogh A, Sonnhammer EL. An HMM posterior decoder for sequence feature prediction that includes homology information. Bioinformatics. 2005 Jun;21 Suppl 1:i251-7. PMID: 15961464.
- Sachdeva G, Kumar K, Jain P, Ramachandran S. SPAAN: a software program for prediction of adhesins and adhesin-like proteins using neural networks. Bioinformatics. 2005 Feb 15;21(4):483-91. PMID: 15374866.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic Local Alignment Search Tool. J Mol Biol 215:403-410; 1990. PMID: 2231712.
- Peters B, Sidney J, Bourne P, Bui HH, Buus S, Doh G, Fleri W, Kronenberg M, Kubo R, Lund O, Nemazee D, Ponomarenko JV, Sathiamurthy M, S choenberger S, Stewart S, Surko P, Way S, Wilson S, Sette A. The immune epitope database and analysis resource: from vision to blueprint. PLoS Biol. 2005 Mar;3(3):e91. PMID: 15760272.
- Peters B, Bui HH, Frankild S, Nielson M, Lundegaard C, Kostem E, Basch D, Lamberth K, Harndahl M, Fleri W, Wilson SS, Sidney J, Lund O, Buus S, Sette A. A community resource benchmarking predictions of peptide binding to MHC-I molecules. PLoS Comput Biol. 2006 Jun 9;2(6):e65. Epub 2006 Jun 9. PMID: 16789818.
- Lin HH, Ray S, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC class I peptide binding prediction servers: applications for vaccine research. BMC Immunol. 2008 Mar 16;9:8. PMID: 18366636.
- Lin HH, Zhang GL, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC-II peptide binding prediction servers: applications for vaccine research. BMC Bioinformatics. 2008 Dec 12;9 Suppl 12:S22. PMID: 19091022.
- Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008 Apr 4;4(4):e1000048. PMID: 18389056.
- De Groot AS, Ardito M, McClaine EM, Moise L, Martin WD. Immunoinformatic comparison of T-cell epitopes contained in novel swine-origin influenza A (H1N1) virus with epitopes in 2008-2009 conventional influenza vaccine. Vaccine. 2009 Sep 25;27(42):5740-7. Epub 2009 Aug 4.PMID: 19660593.
Selected Papers that have Cited Vaxign:
- Caro-Gomez E, Gazi M, Goez Y, Valbuena G. Discovery of novel cross-protective Rickettsia prowazekii T-cell antigens using a combined reverse vaccinology and in vivo screening approach. Vaccine. 2014 Jul 7. pii: S0264-410X(14)00901-3. doi: 10.1016/j.vaccine.2014.06.089. PMID: 25010827.
- Pereira UP, Soares SC, Blom J, Leal CA, Ramos RT, Guimarães LC, Oliveira LC, Almeida SS, Hassan SS, Santos AR, Miyoshi A, Silva A, Tauch A, Barh D, Azevedo V, Figueiredo HC. In silico prediction of conserved vaccine targets in Streptococcus agalactiae strains isolated from fish, cattle, and human samples. Genet Mol Res. 2013 Aug 12;12(3):2902-12. PMID: 24065646.
- Soares SC, Trost E, Ramos RT, Carneiro AR, Santos AR, Pinto AC, Barbosa E, Aburjaile F, Ali A, Diniz CA, Hassan SS, Fiaux K, Guimarães LC, Bakhtiar SM, Pereira U, Almeida SS, Abreu VA, Rocha FS, Dorella FA, Miyoshi A, Silva A, Azevedo V, Tauch A. Genome sequence of Corynebacterium pseudotuberculosis biovar equi strain 258 and prediction of antigenic targets to improve biotechnological vaccine production. J Biotechnol. 2013 Aug 20;167(2):135-41. PMID: 23201561.
- Gomez G, Pei J, Mwangi W, Adams LG, Rice-Ficht A, Ficht TA. Immunogenic and invasive properties of Brucella melitensis 16M outer membrane protein vaccine candidates identified via a reverse vaccinology approach. PLoS One. 2013;8(3):e59751. doi: 10.1371/journal.pone.0059751. Epub 2013 Mar 22. PMID: 23533646. PMCID: PMC3606113.
- Ali A, Soares SC, Santos AR, Guimar?es LC, Barbosa E, Almeida SS, Abreu VA, Carneiro AR, Ramos RT, Bakhtiar SM, Hassan SS, Ussery DW, On S, Silva A, Schneider MP, Lage AP, Miyoshi A, Azevedo V. Campylobacter fetus subspecies: Comparative genomics and prediction of potential virulence targets. Gene. 2012 Oct 25;508(2):145-56. Epub 2012 Aug 6. PMID: 22890137.
- V D'Afonseca, SC Soares, A Ali, AR Santos, Pinto AC, Magalh?es AAC, de Jesus Faria C, Barbosa E, Guimar?es LC, Eslab?o M, Almeida SS, Abreu VAC, Zerlotini A, Carneiro AR, Cerdeira LT, Ramos RTJ, Hirata Jr R, Mattos-Guaraldi AL, Trost E, Tauch A, Silva A, Schneider MP, Miyoshi A, Azevedo V. Reannotation of the Corynebacterium diphtheriae NCTC13129 genome as a new approach to studying gene targets connected to virulence and pathogenicity in diphtheria. Open Access Bioinformatics. 2012:4 1–13. http://dx.doi.org/10.2147/OAB.S25500.
- Jain KK. Synthetic Biology and Personalized Medicine. Med Princ Pract. 2012 Aug 16. PMID: 22907209.
- Oyston P, Robinson K. The current challenges for vaccine development. J Med Microbiol. 2012 Jul;61(Pt 7):889-94. PMID: 22322337. (Review)
- Brumbaugh AR, Mobley HL. Preventing urinary tract infection: progress toward an effective Escherichia coli vaccine. Expert Rev Vaccines. 2012 Jun;11(6):663-76. PMID: 22873125. (Review)
- Kudva IT, Griffin RW, Krastins B, Sarracino DA, Calderwood SB, John M. Proteins other than the locus of enterocyte effacement-encoded proteins contribute to Escherichia coli O157:H7 adherence to bovine rectoanal junction stratified squamous epithelial cells. BMC Microbiol. 2012 Jun 12;12:103. PMID: 22691138.
- Libanova R, Becker PD, Guzmán CA. Cyclic di-nucleotides: new era for small molecules as adjuvants. Microb Biotechnol. 2012 Mar;5(2):168-76. doi: 10.1111/j.1751-7915.2011.00306.x. Epub 2011 Sep 29. PMID: 21958423.
- Jones D. Reverse vaccinology on the cusp. Nat Rev Drug Discov. 2012 Feb 10;11(3):175-6. doi: 10.1038/nrd3679. PMID: 22322255. (News and Analysis)
- He Y, Rappuoli R, De Groot A, Chen RT. Emerging vaccine informatics. Journal of Biomedicine and Biotechnology. 2010 (2010), Article ID 218590, 26 pages. 2010;2010:218590. Epub 2011 Jun 15. PMID: 21772787.
- Moise L, Cousens L, Fueyo J, De Groot AS. Harnessing the power of genomics and immunoinformatics to produce improved vaccines. Expert Opin Drug Discov. 2011 Jan;6(1):9-15. PMID: 22646824. (Review)
- SD Siadat, AS Salmani, MR Aghasadeghi. Brucellosis Vaccines: An Overview. In Book: Zoonosis, edited by Dr. Jacob Lorenzo-Morales. Publisher: InTech. Published online 04, April, 2012. ISBN 978-953-51-0479-7. (Book chapter)
- Thomas C, Moridani M. Interindividual variations in the efficacy and toxicity of vaccines. Toxicology. 2010 Dec 5;278(2):204-10. Epub 2009 Oct 28. PMID: 19837123.
- Tomar N, De RK. Immunoinformatics: an integrated scenario. Immunology. 2010 Oct;131(2):153-68. doi: 10.1111/j.1365-2567.2010.03330.x. Epub 2010 Aug 16. Review. PMID: 20722763.
Note: To find more papers that have cited Vaxign, please check Google Scholar: http://scholar.google.com/scholar?hl=en&q=vaxign.
Useful Web Links:
- AntiJen: A database containing quantitative binding data for peptides binding to MHC Ligand, TCR-MHC Complexes, T Cell Epitope, TAP, B Cell Epitope molecules and immunological Protein-Protein interactions.
- Bcipep: A database of B cell epitopes.
- IEDB Analysis Resource: A list of IEDB epitope prediction and anlaysis tools.
- JenPep: A database of quantitative binding data for immunological protein-peptide interactions.
- RANKPEP: A web-based computational program for prediction of binding peptides to Calss I & II MHC molecules.
|

